- 学科导航
认识儿童肌肉疾病:抗肌萎缩蛋白病
|
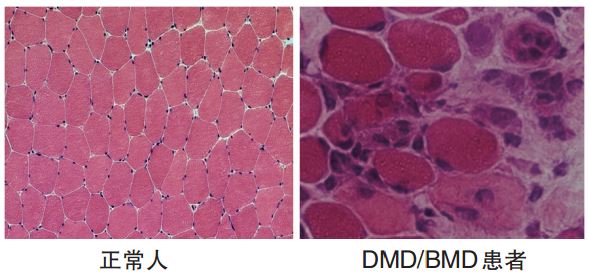
复旦大学附属儿童医院 李西华 抗肌萎缩蛋白病包括Duchenne型肌营养不良(DMD)和Becker型肌营养不良(BMD)。DMD是目前人类已知疾病中最大、最复杂的基因缺陷所导致的,一种随着年龄增加全身肌肉呈进行性消耗和运动功能减退的致死性X伴性隐性遗传性肌肉病,已被列入罕见病范围。患儿通常3~4岁开始步态异常、10~12岁起逐渐丧失行走能力、20~30岁间死于心肺功能衰竭,到目前尚无有效的根治手段。 该病发病率为每3500个新生男婴中就会有1例患DMD者,估计目前国内累计患者已达近10万人。因此,我国已是世界上DMD患者人数最多的国家之一。以下就抗肌萎缩蛋白病的防治进行简要介绍。 DMD的发现及探究 早在1861年,法国医生Duchenne对13例出现进行性肌无力的男性患者进行了详细的描述。患者通常儿童时期起病,肌无力最初累及下肢和腰部肌肉,伴随着某些肌肉的肥大,并用针刺活检的方法对患者的肌肉组织进行分析,发现有大量的结缔组织和脂肪组织增生。在1868年Duchenne发表了“假肥大性或肌硬化性肌肉麻痹” 的权威性论文。 1879年,高尔斯(Gowers)对21例病例进行临床观察,并回顾100个病例的描述。他发现并作图描述了受疾病影响的男孩通过双手支撑在双腿上,从地上站起的特殊方式,即Gowers征。 1955年,德国神经内科医生贝克尔(Becker)和基尔纳(Kiener)描述了一些和DMD具有类似特点的病例,但症状较轻,发病年龄比DMD晚,进展速度更慢。于是Becker提出存在一种良性X连锁肌营养不良,即BMD,并认为DMD和BMD均系同一种病因所引起。 1959年,埃巴特•苏吉托(Ebashi Sugita)发现了DMD/BMD血清肌酸激酶(CK)显著增高,成为诊断本病的生化指标。1973年,杜博维茨(Dubowitz)等提出DMD/BMD的肌肉组织病理学的特征是再生纤维和大量高浓缩肌纤维。 1986年,莫纳科(Monaco)等人采用定位克隆方法分离到抗肌萎缩蛋白(dystrophin)基因得到第1个cDNA片段。1987年凯宁格(Koening)等人又克隆到抗肌萎缩蛋白基因全长的cDNA,同时,刘易斯•孔克尔(Louis Kunkle)研究小组明确了抗肌萎缩蛋白基因缺陷与抗肌萎缩蛋白病的对应关系。 抗肌萎缩蛋白基因跨越2400 kb,其基因组长度约占人基因组的0.1%、X 染色体的1.5%,由79 个外显子和78 个内含子构成,整个基因内含子序列占99%。 抗肌萎缩蛋白基因编码蛋白为427kDa ,位于肌细胞膜的内层肌细胞膜磷脂双层结构中,连接着胞外细胞外基质(ECM)和胞内丝状肌动蛋白(F-actin)组成的细胞骨架蛋白,与肌营养不良蛋白聚糖(dystroglycans)、肌聚糖蛋白(sarcoglycans)和互养蛋白(syntrophins)三种糖蛋白共同构成了多分子复合物DGC(dystrophin associated glycoprotein complex)。在这个复合物中, 抗肌萎缩蛋白对维持肌细胞膜的完整性、肌力的产生和传递起着关键的作用。当抗肌萎缩蛋白基因发生缺陷,使肌细胞膜下抗肌萎缩蛋白不同程度的丢失,则会导致骨骼肌纤维变性、坏死和再生为主要特点的病理改变(图1)。
如何发现DMD征兆 如有下列3个表现,可怀疑(即使无DMD家族史)。 1.肌肉功能问题 通常是家人先发现异常,男童如患DMD,走路比同龄男童较晚,腓肠肌肥大,跑步、跳跃和爬楼梯均有困难,走路容易跌倒,可能有踮起脚尖走路的趋势,说话也可能较晚。DMD患儿典型特征之一是Gowers征,由于腰部和大腿肌肉首先受损,男童从地上爬起时,必须先用手撑地,然后借助手臂之力挺起腰部,但是Gowers征也可在其他类型的肌病中见到。 2.血液中的肌酸激酶(CK)水平升高 发现CK水平明显升高应及时转诊到神经肌肉病专科进行明确诊断。高水平的CK也能在其他种类肌肉疾病的患者中见到,单凭高CK不足以确诊DMD。 3.血液中的肝酶如天冬氨酸氨基转移酶(AST)和丙氨酸氨基转移酶(ALT)水平升高 血液中这些酶的水平升高通常与肝病有关,但是肌营养不良也能引起它们的升高。因此,肝酶升高但不伴有明显肝病症状时,不要立即进行肝脏活检,应首先怀疑肌营养不良的可能。 如何诊断 1.基因检测 多重连接探针扩增技术(MLPA)是目前对DMD基因检测首选的方法,它覆盖了DMD基因79个外显子,能够检出基因缺失和重复两种突变类型。如果MLPA检测为阴性结果时,可进一步采用肌肉活检的方法明确诊断,以防误诊。 2.肌肉活检 在麻醉状态下,外科医生切取患儿绿豆粒大小样的肌肉组织(通常选择肱二头肌),对此标本进行分析。在正常人肌纤维的肌膜上可看见完整的、连续的、强度均匀的环形荧光条带围绕在每根肌纤维上,说明抗肌萎缩蛋白完整存在;在DMD患者肌肉中没有看到环形的红色荧光条带,说明蛋白已经完全缺失;在BMD患者肌肉中可以看到连续或断续的、较弱的环形红色荧光条带,与正常人比较,抗肌萎缩蛋白表达程度明显减弱,说明部分蛋白已缺失,还残留一部分蛋白维持肌肉的功能。 遗传咨询 有时候引起DMD基因突变是随机的,称为自发突变。有的病例还可能与母亲遗传有关。如果母亲存在基因突变,她就是个“携带者”,她把基因突变传给孩子,男孩就有50%概率会患有DMD,而女孩则有50%概率为“携带者”。 如果母亲接受了基因检测找到了突变类型,她在未来妊娠前就应先接受遗传咨询,为产前诊断做好准备,并且母亲家族中有血缘关系的女性成员也应接受检测,把生育DMD男孩的风险降到最低。 治疗方案 长期服用肾上腺皮质激素(以下简称激素)加上康复治疗能延缓病情的发展。激素是目前能减慢DMD 孩童肌力和运动功能衰退的唯一药物。其目的是帮助孩子延长走路时间,提高活动参与性,同时也减少呼吸、心脏和骨骼问题。使用激素前及长期口服中要做好各种预防措施,如定期随访血压、电解质等,以避免药物的副作用。 服用激素最佳时间是孩子运动功能处在平台期(一般在4-6岁),平台期是指孩子的活动能力停止改善而又没有明显的衰退阶段。通常2岁以下的DMD孩童不推荐使用激素,在决定服用激素前应确认完成各种疫苗的接种。 开始服用激素时,建议每天早上服用,按 0.75mg/(kg•d)剂量饭前口服泼尼松。若孩子在早上服用药物后数小时,出现短暂的副作用如过度活跃或情绪不稳,可考虑改在下午服药,可能会缓解一些不良反应。 来源:中国医学论坛报 |
 收藏
收藏

热门新闻
- 阅读
- 评论




推荐阅读
-
儿童感染后用什么药?哪些情况需要就医?北

儿童感染新冠病毒的症状、病程有何特点?退烧药怎么选、怎么吃?孩子出现何种症状需立[详细]
-
国务院最新公布:我国医养结合面临问题和下

导语医养结合将成为下一个风口!在日前召开的第十三届全国人大常委会第三十六次会议上[详细]

-
版权所有:北京英芙麦迪科技有限公司
地址:北京市顺义区竺园二街2号院8号楼301(天竺综合保税区)
电话:(010)-80489293-6011 邮件:hmp@bjhanmi.com.cn
-
 互联网药品信息服务资格证书 京ICP备07502511号-4
互联网药品信息服务资格证书 京ICP备07502511号-4药品医疗器械网络信息服务备案 (京)网药械信息备字(2022)第00010号
(京)-非经营性-2019-0001
 京公网安备11011302001972
京公网安备11011302001972